Network and Enrichment Analysis
Last updated on 2025-11-14 | Edit this page
Overview
Questions
- How can DEA results be used to identify biologically meaningful patterns?
- How can we visualise and interpret protein–protein interaction (PPI) networks and pathway enrichment results?
Objectives
- Visualise significant proteins using network analysis (STRING)
- Identify enriched biological processes and pathways (GO and KEGG)
Now that we have a list of differentially expressed (DE) proteins, we can explore how these proteins interact with each other and what biological functions or pathways they are involved in.
This step helps translate statistical significance into biological meaning by integrating our results with known protein–protein interactions (PPIs) and functional annotation databases.
STRING protein–protein interaction network analysis
The STRING database integrates known and predicted PPIs from multiple sources (e.g. experimental data, text mining, prediction methods, and curated databases).
We can visualise DE proteins within the context of their interaction network to identify clusters or modules potentially corresponding to biological pathways.
R
# Create an instance of the STRINGdb reference class for humans (species 9606)
string_db <- STRINGdb$new(version = "11.5", species = 9606, score_threshold = 400)
# Map STRING identifiers to gene identifiers in our DEA results dataframe
mapped <- string_db$map(results, "Protein.Names", removeUnmappedRows = TRUE)
# Only map the proteins of significance
de_proteins_mapped <- mapped %>% filter(adj.P.Val < 0.05)
# Plot network of significant proteins
string_db$plot_network(de_proteins_mapped$STRING_id)
Note: if the above code takes too long to load, you can download
an RData that contains the string_db and
mapped functions here.

How many clusters are there?
How many of these interactions are predicted versus experimentally validated? (See image below for legend)
You can explore how these proteins interact in an interactive setting by going to https://string-db.org/ and further exploring your proteins of interest.

More data exploration using STRINGdb
There are many ways of exploring your data using the STRING database - too many to cover in one tutorial!
You can learn more by reading the vignette
or inspecting available functions within the STRINGdb
package by running:
R
STRINGdb$methods()
OUTPUT
[1] ".objectPackage" ".objectParent"
[3] "add_diff_exp_color" "add_proteins_description"
[5] "benchmark_ppi" "benchmark_ppi_pathway_view"
[7] "callSuper" "copy"
[9] "enrichment_heatmap" "export"
[11] "field" "get_aliases"
[13] "get_annotations" "get_bioc_graph"
[15] "get_clusters" "get_enrichment"
[17] "get_graph" "get_homologs"
[19] "get_homologs_besthits" "get_homology_graph"
[21] "get_interactions" "get_link"
[23] "get_neighbors" "get_paralogs"
[25] "get_pathways_benchmarking_blackList" "get_png"
[27] "get_ppi_enrichment" "get_ppi_enrichment_full"
[29] "get_proteins" "get_pubmed"
[31] "get_pubmed_interaction" "get_subnetwork"
[33] "get_summary" "get_term_proteins"
[35] "getClass" "getRefClass"
[37] "import" "initFields"
[39] "initialize" "load"
[41] "load_all" "map"
[43] "mp" "plot_network"
[45] "plot_ppi_enrichment" "post_payload"
[47] "ppi_enrichment" "remove_homologous_interactions"
[49] "set_background" "show"
[51] "show#envRefClass" "trace"
[53] "untrace" "usingMethods" Szklarczyk, D., Nastou, K., Koutrouli, M., Kirsch, R., Mehryary, F., Hachilif, R., Hu, D., Peluso, M. E., Huang, Q., Fang, T., Doncheva, N. T., Pyysalo, S., Bork, P., Jensen, L. J., & von Mering, C. (2025). The STRING database in 2025: protein networks with directionality of regulation. Nucleic acids research, 53(D1), D730–D737. https://doi.org/10.1093/nar/gkae1113
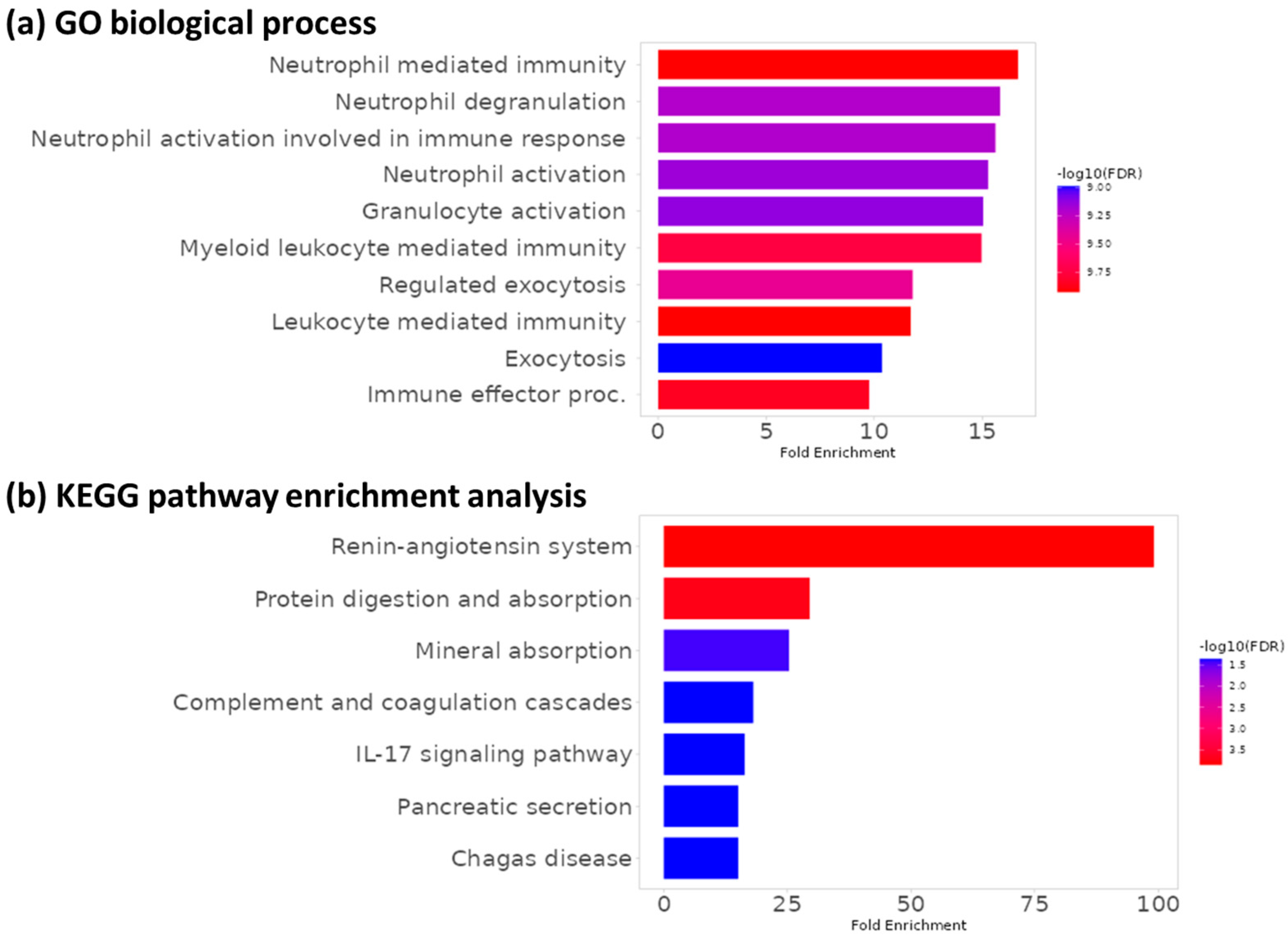
Functional enrichment analysis (GO and KEGG)
Using the significant DE proteins, we can perform functional enrichment analysis to determine whether certain biological processes (BP), cellular components (CC), or molecular functions (MF) are over-represented.
Two common types of enrichment exploration are:
- Gene Ontology (GO): describes BPs, CCs, amd MFs
- KEGG pathways: curated molecular interactions
GO enrichment analysis
R
#Subset genes from results table
de_proteins <- results %>% filter(adj.P.Val < 0.05)
# Convert UniProt IDs to Entrez IDs for enrichment analysis
converted <- bitr(de_proteins$Protein.Group,
fromType = "UNIPROT",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
OUTPUT
'select()' returned 1:1 mapping between keys and columnsWARNING
Warning in bitr(de_proteins$Protein.Group, fromType = "UNIPROT", toType =
"ENTREZID", : 7.69% of input gene IDs are fail to map...R
# GO Biological Process enrichment
ego <- enrichGO(gene = converted$ENTREZID,
OrgDb = org.Hs.eg.db,
ont = "ALL", # looking at all 3 subontologies
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
readable = TRUE)
# Show plot
dotplot(ego, showCategory = 10)

KEGG pathway enrichment analysis
R
# KEGG pathway enrichment
ekegg <- enrichKEGG(gene = converted$ENTREZID,
organism = 'hsa',
pvalueCutoff = 0.05)
OUTPUT
Reading KEGG annotation online: "https://rest.kegg.jp/link/hsa/pathway"...OUTPUT
Reading KEGG annotation online: "https://rest.kegg.jp/list/pathway/hsa"...R
dotplot(ekegg, showCategory = 10)

{kind=link}
While there are many similar pathways, the exact pathways are not identical. This is likely due to the fact we are using a subset of the larger dataset in this tutorial, so we have a slightly different list of DE proteins.
- Differential expression analysis identifies statistically significant changes in protein abundance between conditions, but does not necessarily tell us the biological significance of such identified proteins.
- Network analysis (STRING) can reveal physical or functional interactions among DE proteins.
- Enrichment analysis (GO/KEGG) can link those proteins to known biological processes or pathways.
- Using multiple approaches can help you interpret proteomics data to measure reliability and consistency of results.