Content from Exploring the dataset
Last updated on 2025-11-14 | Edit this page
Overview
Questions
- What is the data we are using in this tutorial?
Objectives
- Understand the dataset we are using in this tutorial.
- Learn how to download publicly available proteomics data from within R.
The data
The data we are using in this tutorial comes from a study done exploring a non-invasive diagnostic approach for Inflammatory Bowel Disease (IBD) using mass spectrometry and machine learning to analyse the stool proteome. Traditional diagnostic methods like colonoscopies are invasive, prompting the need for alternatives. The researchers collected 123 stool samples and identified 48 differentially expressed proteins, narrowing them down to 7 key proteins using feature selection. A Support Vector Machine (SVM) model was developed, achieving 96% sensitivity and 76% specificity in distinguishing active IBD patients from symptomatic non-IBD patients. This approach demonstrates the potential for accurate, non-invasive IBD diagnosis, improving patient management and reducing the need for invasive procedures.
For the purpose of this tutorial, we will be using a subset of the total data presented in the paper, including:
-
11 control (Ctrl) samples
- 6 from batch one + 5 from batch three
-
9 active Crohn’s disease (aCD) samples
- 5 from batch one + 4 from batch three
Reference:
Shajari, E.; Gagné, D.; Malick, M.; Roy, P.; Noël, J.-F.; Gagnon, H.; Brunet, M.A.; Delisle, M.; Boisvert, F.-M.; Beaulieu, J.-F. Application of SWATH Mass Spectrometry and Machine Learning in the Diagnosis of Inflammatory Bowel Disease Based on the Stool Proteome. Biomedicines 2024, 12, 333. https://doi.org/10.3390/biomedicines12020333
Challenge
Can you see where to find the publicly available data in the paper?
Publicly available data can be found under the ‘Data Availability Statement’ in the paper.
Loading the data
Let’s start by loading the packages required to access data stored on PRIDE.
PRIDE is part of the ProteomeXchange Consortium, which provides open access to proteomics data.
For more background on navigating PRIDE data, you can look through this course: PRIDE Quick Tour (EMBL-EBI Training)
R
library(rpx)
library(dplyr)
The rpx package gives us programmatic access to the
ProteomeXchange without having to leave R.
R
# Set the dataset identifier and load
px_id <- 'PXD047585'
px <- PXDataset(px_id)
We can then perform several functions to extract the contents of the
px object:
R
# Print the dataset title
pxtitle(px)
OUTPUT
[1] "Application of SWATH Mass Spectrometry and Machine Learning in Diagnosis of Inflammatory Bowel Disease Based on Stool Proteome"R
# Print a link to the dataset on PRIDE
pxurl(px)
OUTPUT
[1] "ftp://ftp.pride.ebi.ac.uk/pride/data/archive/2024/02/PXD047585"R
# Print the taxonomy (organism) information for the dataset
pxtax(px)
OUTPUT
[1] "Homo sapiens (human)"Exploring dataset files
Let’s see which files are included in this dataset.
R
# Retrieve a list of dataset files
px_files <- pxfiles(px)
R
# Display the first few files
head(px_files)
OUTPUT
[1] "20201016_UdSjfb_20190115_freshstool_003.mzML"
[2] "20201016_UdSjfb_20190115_freshstool_003.wiff"
[3] "20201016_UdSjfb_20190115_freshstool_003.wiff.scan"
[4] "20201016_UdSjfb_20190115_freshstool_005.mzML"
[5] "20201016_UdSjfb_20190115_freshstool_005.wiff"
[6] "20201016_UdSjfb_20190115_freshstool_005.wiff.scan"PRIDE datasets often include a mix of raw data, search results, processed outputs, and metadata.
To understand the data composition, we’ll examine file extensions.
R
# Count the number of files by extension
table(sapply(strsplit(px_files, "\\."), tail, 1))
OUTPUT
fas mzML pdf scan speclib tsv txt wiff xlsx
1 78 1 78 2 13 1 78 3 Alternatively, we can get the same result with the tools package:
R
library(tools)
table(file_ext(px_files))
OUTPUT
fas mzML pdf scan speclib tsv txt wiff xlsx
1 78 1 78 2 13 1 78 3 Each file extension represents a specific proteomics data type. For
example, .raw may indicate vendor instrument output and
.mzML indicates an open format for MS data.
For more information about different proteomics file formats, check out:
Challenge
Try visiting the dataset page on the PRIDE website and compare:
The number of files listed there versus those returned by rpx
The types of files (e.g., .raw, .mzML, .txt, etc.)
Questions to consider:
Why might the PRIDE web interface and R output differ?
What are the advantages of using programmatic access via rpx?
Programmatic access allows you to automate each step of your analysis, making your workflow fully reproducible.
Downloading required files
Good news! We have already downloaded and processed the data for you, as described in the next lesson. You should have downloaded this from the Setup page.
If you wish to download files from another dataset, you can use the
curl package as demonstrated below.
R
library(curl)
curl::curl_download(url='https://ftp.pride.ebi.ac.uk/pride/data/archive/2024/02/PXD047585/SampleAnnotation.xlsx',destfile = 'data/SampleAnnotation.xlsx')
Note:
-
We retrieved the
urlin an earlier step usingpxurl().- We have replaced
ftp://at the start withhttps:// - We have specified the name of the file we wish to download
(
SampleAnnotation.xlsx), from the list we sourced earlier usingpxfiles()
- We have replaced
We set
destfileto our desired name and location for the downloaded file.
- The dataset used in this tutorial includes stool samples from a study investigating non-invasive diagnostic methods for Inflammatory Bowel Disease.
- We can use the
rpxpackage to download and explore data from PRIDE or the ProteomeXchange Consortium without leaving R.
Content from Processing data with DIA-NN
Last updated on 2025-11-14 | Edit this page
Overview
Questions
- How can I process my output files from a mass spectrometry-based proteomics experiment?
Objectives
- Learn how to process proteomics data using DIA-NN.
Processing proteomics data
There are many software options for processing mass spectrometry-based proteomics output. These software match mass spectra against an empirical or predicted spectral library to identify peptides and infer the protein to which they belong.
Some software you may have heard of include Spectronaut, Fragpipe, MaxQuant and DIA-NN. Each software have slightly different functionality and attributes, and are frequently updated. In this tutorial, we are demonstrating the use of DIA-NN, a software designed to process DIA proteomics data.

Why DIA-NN?
Key features of DIA-NN software include:
- Neural network–based scoring: Improves identification confidence.
- Interference correction: Enhances quantification accuracy.
- Library-free analysis: Can work without a spectral library (generating one directly from DIA data).
- High speed and efficiency: Suitable for large-scale datasets.
- Cross-run normalisation: Ensures consistent quantification across experiments.
- Command-line executable: DIA-NN can be run from a graphical user interface (GUI) or directly from the command line, making it easy to integrate into an automated pipeline.
DIA-NN is free to download. Detailed operating instructions and a link to install the latest version of DIA-NN can be found on the GitHub page.
DIA-NN workflow
Important Note
Due to the amount of time it will take to process the data, you do not need to install or operate DIA-NN for the purposes of this tutorial. Pre-processed data is provided, which has been prepared following the procedures described below.
Part 1: Creating a spectral library
DIA-NN analysis consists of two steps. The first step is to generate a predicted spectral library for your experimental organism.
-
Download a sequence database from UniProt.
Select the relevant
organismfor your sample andReviewed (Swiss-Prot)status to filter results.Click
DownloadChange the format to
FASTA (canonical and isoform)Select
Nounder Compressed and download the FASTA file
FOR SPECIFIC SEARCHES
If you need to search for a variant protein or peptide that differs from the standard organism proteome, you can manually edit the protein sequences in the UniProt FASTA using a text editor.
Download a contaminant FASTA file. For most experiments, the Universal Contaminants.fasta will be appropriate, but you may prefer to use one of the sample-type specific contaminant FASTAs depending on your experiment. More information about this resource and contaminants will be discussed in the next lesson.
Open the DIA-NN GUI or a command line interface. If there are multiple versions of DIA-NN installed on your device, ensure to update
DIA-NN exewith the file path to the version you wish to use.Add the UniProt and Contaminant FASTA files you just downloaded.
At the time of writing, DIA-NN 2.2.0 cannot read or write files saved in an external drive. Please ensure all input and output files are saved on a local drive to avoid error.
Examples are included below for the additional settings we recommend changing from the default. Depending on your experiment and research goals, you may wish to change additional settings from the default - read more in the DIA-NN documentation.
Run. DIA-NN will output a predicted spectral library in the
.speclibfile type.

BASH
C:\DIA-NN\2.2.0\diann.exe ` # Replace with file path to DIA-NN on your device
--lib "" `
--out "C:\path\to\output\report.parquet" ` # Replace with desired file path to output report.parquet
--out-lib "C:\path\to\output\report-lib.parquet" ` # Replace with desired file path to output report-lib.parquet
--fasta "C:\path\to\UniprotFasta\.fasta" ` # Replace with file path to Uniprot FASTA
--fasta "C:\path\to\ContaminantsFasta\.fasta" ` # Replace with file path to contaminants FASTA
--threads 30 ` # Replace with the number of threads to use for this analysis
--verbose 1 `
--qvalue 0.01 `
--matrices `
--gen-spec-lib `
--predictor `
--reannotate `
--fasta-search `
--min-fr-mz 200 `
--max-fr-mz 1800 `
--met-excision `
--min-pep-len 7 `
--max-pep-len 30 `
--min-pr-mz 300 `
--max-pr-mz 1800 `
--min-pr-charge 1 `
--max-pr-charge 4 `
--cut K*,R* `
--missed-cleavages 1 `
--unimod4 `
--rt-profiling This spectral library can be re-used for all analyses of samples from the same organism. It is recommended to re-download the UniProt FASTA and re-run this step approximately once a month to ensure your library is up-to-date.
Part 2: Analysis
The second step is to search your raw files (mass spectrometry output files) against the spectral library.
Open the DIA-NN GUI or a command line interface. If there are multiple versions of DIA-NN installed on your device, ensure to update
DIA-NN exewith the file path to the version you wish to use.-
Upload your raw files.
- If using the GUI, under
InputselectRawthen select all of the files you wish to search. - If using the command line, you can use
--fmultiple times to list each file individually, or (recommended) use--dir [folder]to instruct DIA-NN to process all raw files saved in a particular folder.
- If using the GUI, under
Upload your spectral library.
Empirical vs predicted spectral library
You may wish to generate an empirical spectral library from your data, and use this for future analyses. An empirical library will be smaller than a predicted library and therefore increase the speed of analysis; however, may result in fewer identifications. We only recommend using an empirical library if generated from high quality data.
You can generate an empirical library from your samples by selecting Generate spectral library and specifying the location of its output.
-
Adjust settings according to your experiment and research goals. Again, examples are included below for the additional settings we recommend changing from the default. You may wish to change additional settings from the default - read more in the DIA-NN documentation.
- In this example, the protease Trypsin was used to generate peptides during sample preparation.
- Leaving Mass accuracy and MS1 accuracy on the default 0 means DIA-NN will optimise these parameters automatically for the first run in the experiment and then reuse the optimised settings for other runs. The DIA-NN documentation recommends different settings depending on the type of mass spectrometer used to generate your data. In this example, data were generated on a TripleTOF 6600, so we set Mass accuracy to 20.0 and MS1 accuracy to 12.0
- Set the number of Threads to be no more than the number of logical cores on your computer.
- In this experiment, we have set the maximum number of variable modifications to 0; however, it is common for this setting to be relaxed in the range of 1 - 3.
- DIA-NN’s default setting is to activate match-between-runs. From the documentation: In MBR mode, DIA-NN does two passes over the data. During the first pass, it creates an empirical spectral library from the data. During the second pass, it reanalyses the experiment with this empirical library, which may result in much improved identification numbers, data completeness and quantification accuracy.

BASH
C:\DIA-NN\2.2.0\diann.exe ` # Replace with file path to DIA-NN on your device
--dir "C:\path\to\folder\of\raw\files" ` # Replace with file path to folder containing all raw files to be analysed
--lib "C:\path\to\SpectralLibrary\report-lib.predicted.speclib" ` # Replace with file path to spectral library
--out "C:\path\to\output\report.parquet" ` # Replace with desired file path to output report.parquet
--threads 30 ` # Replace with the number of threads to use for this analysis
--verbose 1 `
--qvalue 0.01 `
--matrices `
--unimod4 `
--mass-acc 20 `
--mass-acc-ms1 12.0 `
--reanalyse `
--rt-profiling Challenge
If you want to run DIA-NN from the command line but are unsure how to write the code, you can update your settings in the GUI, click ‘Run’, and copy the commands output in the log. That’s how we got the example code above!
Can you match each line of code to the corresponding setting in the GUI?
Check the description of available commands in the DIA-NN documentation
DIA-NN output
DIA-NN output includes several files, including:
| Output file | Description |
|---|---|
| log.txt | A log of what DIA-NN has run. You should always use
ctrl + F to check for any ‘ERROR’ or ‘WARNING’ messages
issued by DIA-NN during your run. |
| .parquet | This is what we will be using for subsequent analyses in this tutorial. |
| pg_matrix.tsv | Matrix of protein values generated by DIA-NN. |
| pr_matrix.tsv | Matrix of peptide values generated by DIA-NN. |
| trends.pdf | A PDF report listing the number of peptides, proteins, and other quality control measures detected for each sample. |
Challenge
If DIA-NN is run through the command line rather than the GUI, the trends.pdf and runs.pdf reports are not automatically generated. Using the DIA-NN documentation, can you work out how to generate this report via the command line?
We have now processed our raw files, identifying the peptides present in our samples and inferring the proteins to which they belong.
For more background information, see the original publication:
Demichev, V., et al. (2020). DIA-NN: neural networks and
interference correction enable deep proteome coverage in high
throughput. Nature Methods. https://doi.org/10.1038/s41592-019-0638-x.
For further details about the different DIA-NN settings, see the documentation.
If you come across any issues, the creators are fairly active at responding to issues and discussions on their GitHub.
- Raw files from a mass spectrometry-based DIA proteomics experiment can be processed using DIA-NN (peptide identification and protein inference).
- The DIA-NN workflow has two key steps: generating a predicted spectral library, and processing raw files.
- The DIA-NN documentation contains detailed information about changing default settings as appropriate for your experiment.
Content from Data cleaning with R
Last updated on 2025-11-14 | Edit this page
Overview
Questions
- How do we convert our processed proteomics data (DIA-NN output) into an analysis-ready format?
Objectives
- Understand the steps required to clean proteomics data and generate
analysis-ready protein values using
limpa.
Load processed data and sample annotation
After processing raw proteomics data, further cleaning and analysis can be conducted using R, Python, or by importing data to other platforms like Perseus or Skyline.
In this tutorial, we will be teaching you one method of cleaning and analysing proteomics data using R. To follow along the remainder of this tutorial, you should open a new R script in RStudio and run the example code provided.
The limpa package
In this tutorial, we are using the limpa package to load
and clean our data. This package was published in 2025 by the Smyth Lab, based at the
Walter and Eliza Hall Institute of Medical Research (WEHI).
For background information, see the original publications:
Li, M., Cobbold, S. A., & Smyth, G. K. (2025). Quantification and differential analysis of mass spectrometry proteomics data with probabilistic recovery of information from missing values. bioRxiv, 2025.2004.2028.651125. https://doi.org/10.1101/2025.04.28.651125
Li, M., & Smyth, G. K. (2023). Neither random nor censored: estimating intensity-dependent probabilities for missing values in label-free proteomics. Bioinformatics, 39(5). https://doi.org/10.1093/bioinformatics/btad200
You can also find further information in the vignette or reference manual accessed via the Bioconductor page.
Setup
First, load the required R packages.
R
library(limpa)
library(readxl)
library(pheatmap)
library(EnhancedVolcano)
library(STRINGdb)
library(clusterProfiler)
library(org.Hs.eg.db)
library(arrow)
library(rpx)
library(dplyr)
library(stringr)
Load DIA-NN output
Load the peptide-level data processed by DIA-NN
(.parquet file), filtering observations by
quality metrics as recommended in the limpa
vignette.
The peptide values will automatically be converted to log2 upon import.
R
y.peptide <- readDIANN(file='data/MBIntroToProteomics.parquet',format="parquet",
q.columns = c("Q.Value","Lib.Q.Value","Lib.PG.Q.Value"),
q.cutoffs = 0.01)
OUTPUT
Length of q-value columns does not match with length of q-value cutoffs. Use q.cutoffs[1] for all columns.Challenge
These quality filtering metrics are recommended in the limpa vignette for data searched in DIA-NN with match-between-runs.
What q.columns are recommended for data searched
without MBR?
R
y.peptide <- readDIANN(file='data/MBIntroToProteomics.parquet',format="parquet",
q.columns = c("Q.Value","Global.Q.Value","Global.PG.Q.Value"),
q.cutoffs = 0.01)
Let’s look at the structure of the data.
R
names(y.peptide)
OUTPUT
[1] "E" "genes"R
head(y.peptide$E)
head(y.peptide$genes)
As you can see, our data has been imported as a limma EList object, with components ‘E’ (log2 peptide matrix) and ‘genes’ (feature annotation) containing information about each peptide.
Load sample annotation
We also need to import information about our samples for later analyses.
R
samples_stool <- as.data.frame(readxl::read_excel('data/SampleAnnotation.xlsx'))
head(samples_stool)
OUTPUT
file_order Sample_id-new Batch Class
1 33 B1_29_c B1 CDr
2 93 B1_87_e B1 UCr
3 71 B1_66_e B1 UCr
4 9 B1_08_b B1 aCD
5 26 B1_22_c B1 CDr
6 31 B1_27_c B1 CDr
file_name
1 D:\\Elmira\\IBD\\01-MSconverter\\00-DIA_NN input-3 batches\\mix.mzMLpeakpicking(batch 1,2)\\Mix\\20201016_UdSjfb_20190115_freshstool_067-29.mzML
2 D:\\Elmira\\IBD\\01-MSconverter\\00-DIA_NN input-3 batches\\mix.mzMLpeakpicking(batch 1,2)\\Mix\\20201016_UdSjfb_20190115_freshstool_187.mzML
3 D:\\Elmira\\IBD\\01-MSconverter\\00-DIA_NN input-3 batches\\mix.mzMLpeakpicking(batch 1,2)\\Mix\\20201016_UdSjfb_20190115_freshstool_143-66.mzML
4 D:\\Elmira\\IBD\\01-MSconverter\\00-DIA_NN input-3 batches\\mix.mzMLpeakpicking(batch 1,2)\\Mix\\20201016_UdSjfb_20190115_freshstool_019-8.mzML
5 D:\\Elmira\\IBD\\01-MSconverter\\00-DIA_NN input-3 batches\\mix.mzMLpeakpicking(batch 1,2)\\Mix\\20201016_UdSjfb_20190115_freshstool_053-22.mzML
6 D:\\Elmira\\IBD\\01-MSconverter\\00-DIA_NN input-3 batches\\mix.mzMLpeakpicking(batch 1,2)\\Mix\\20201016_UdSjfb_20190115_freshstool_063-27.mzMLWe need to clean our sample annotation data to add an identifying column which matches the sample column names in our peptide matrix.
Given we are only using a subset of the main dataset in this tutorial, we can also filter the sample annotation dataframe to only include rows pertaining to the samples in our subset.
R
# Create sample_name column that matches peptide matrix column names
samples_stool$fixed_file_name <- gsub("\\\\", "/", samples_stool$file_name)
samples_stool$fixed_file_name <- basename(samples_stool$fixed_file_name)
samples_stool$sample_name <- gsub(".mzML", "",samples_stool$fixed_file_name)
# Filter to only samples present in our dataset
samples_stool <- samples_stool %>% filter(samples_stool$sample_name %in% colnames(y.peptide$E))
We can attach our sample annotation data to the EList object for later analyses.
R
# Ensure sample row order matches column order of the peptide matrix
sample_info <- samples_stool[samples_stool$sample_name %in% colnames(y.peptide$E),]
rownames(sample_info) <- sample_info$sample_name
y.peptide$E <- y.peptide$E[,rownames(sample_info)]
# Add experimental metadata to EList
y.peptide$targets <- sample_info[, c('Class', 'Batch')]
Quality filtering
After reading in the data, it is common to conduct additional filtering to improve the quality of your data and assist with downstream interpretation of results.
- Filtering out non-proteotypic peptides: Removes peptides that belong to more than one protein.
- Filtering out compound proteins: Removes peptides belonging to compound protein groups consisting of multiple proteins separated by “;” delimiters.
- Filtering out singleton peptides: Removes peptides that are the only peptide belonging to that protein (i.e. keeps only proteins with multiple peptides).
R
y.peptide <- filterNonProteotypicPeptides(y.peptide)
y.peptide <- filterCompoundProteins(y.peptide)
y.peptide <- filterSingletonPeptides(y.peptide, min.n.peptides = 2)
These filtering steps are optional; if they are not run,
limpa will still be able to conduct protein quantification.
For more information about these peptide-level filters and whether they
are appropriate for your experiment, please consult the limpa
documentation.
Removing contaminants
Contaminant background signals can influence mass spectrometry-based proteomics data. It is essential to remove contaminant proteins from the sample preparation process to avoid confounding results.
Frankenfield et al. (2022) created a new set of sample-type specific and universal contaminant FASTA files which they showed reduced false identifications, increased protein identifications and did not influence protein quantification for DIA workflows. These FASTA files updated upon widely used contaminant protein lists from MaxQuant and the common Repository of Adventitious Proteins (cRAP).
In the previous lesson, we incorporated the universal contaminants FASTA into our spectral library. This FASTA is structured such that all contaminant proteins are named beginning with ‘Cont_’, which means we can simply remove any proteins from our analysis containing this string text in their name.
R
# Remove contaminant proteins from the genes dataframe
y.peptide$genes <- y.peptide$genes %>% filter(!str_detect(Protein.Group, "Cont_"))
# Match peptides in the matrix to only those remaining in the dataframe
y.peptide$E <- y.peptide$E[rownames(y.peptide$genes), ]
More about contaminants
You can download the contaminant FASTA files from GitHub.
If you are analysing data that has been processed without a contaminant FASTA, you can also download an Excel spreadsheet contaminant list from the same link and use this to identify and remove contaminant proteins from your dataset.
For more information, see the original publication:
Frankenfield, A. M., Ni, J., Ahmed, M., & Hao, L. (2022). Protein Contaminants Matter: Building Universal Protein Contaminant Libraries for DDA and DIA Proteomics. Journal of proteome research, 21(9), 2104–2113. https://doi.org/10.1021/acs.jproteome.2c00145
Protein quantification
Missing data has been a longstanding issue impacting the accuracy and reproducibility of mass spectrometry-based proteomic analyses, complicating downstream analyses. Most established imputation methods are not designed to account for the specific attributes of proteomics data, and there is no consensus in the literature for how missing values should be managed in proteomics experiments. This presents an issue with using the protein values directly generated by platforms like DIA-NN, which contain many missing values.
In this tutorial, we are using the dpcQuant() method of
protein quantification from the limpa package, or
Linear Models for Proteomics Data (Li, Cobbold &
Smyth, 2025). The detection probability curve (DPC)
models the relationship between peptide intensity and missingness,
showing that almost all missing values in label-free proteomics data are
not missing at random and should be accounted for (Li & Smyth,
2023). The dpcQuant() uses Bayesian statistical methods
based on peptide intensities and the DPC coefficients for a particular
dataset to generate a complete matrix of protein values without the need
for imputation. This method outputs a linear model object suitable for
downstream analysis with the limma package.
To learn more about the detection probability curve and the
limpa package, please read the original publications.
Using limpa and the DPC for protein quantification
First, we will generate and visualise our detection probability curve.
The DPC coefficients are related to the dataset and software used for processing. A steeper slope means there is more statistical information in the pattern of missing data. A slope closer to 0 means there is more data missing at random.
R
# Estimate the DPC for our data
dpcfit <- dpcON(y.peptide, robust=TRUE)
# Print the intercept and slope of our DPC
dpcfit$dpc
OUTPUT
beta0 beta1
-7.2678650 0.6033267 R
# Plot the DPC
plotDPC(dpcfit)

Next, we can use the DPC to generate our log2 protein values. This function requires the following input:
- a limma EList object, or a numeric matrix, of peptide-level log2
expression values, with samples as columns and peptides as rows
(
y.peptide) - vector of protein IDs (
"Protein.Names") - numeric vector providing the slope and intercept of the DPC
(
dpcfit)
R
y.protein <- dpcQuant(y.peptide, "Protein.Names", dpc=dpcfit)
OUTPUT
Estimating hyperparameters ...OUTPUT
Quantifying proteins ...OUTPUT
Proteins: 280 Peptides: 2672Note: our dataset is quite small, so this process should only take a few seconds. For larger datasets, it may take minutes or hours for this command to run.
Alternate versions of the DPC
The base dpc() assumes observed log-intensities are
normally distributed.
A modified version, used in this tutorial,
dpcON(robust=TRUE) downweighs outliers.
Alternatively, dpcCN() assumes the complete set of
log-intensities are normally distributed. Using this method may
underestimate the DPC slope if there is a non-small proportion of
peptides missing by chance.
You can also choose your own slope by passing the argument
dpc.slope= to dpcQuant() instead of
dpc=dpcfit. Using this method, an intercept value will
automatically be generated. You can check this value by using
estimateDPCIntercept(). The authors recommend a slope of
0.6 - 0.8 is usually appropriate for data generated using DIA-NN.
You may wish to conduct further statistical analyses to determine which method is most appropriate for your experiment; however, the authors suggest that downstream analysis should not be significantly affected by differing DPC coefficients.
You can read more about the differences between each version of the DPC in the paper or the limpa manual.
Challenge
The limpa package also provides a function to impute missing peptide values and generate a complete peptide matrix using the DPC. Can you search the limpa documentation to identify the command used to impute and quantify missing values in a rowwise manner?
dpcImpute()
dpcQuant() output
We have now generated an EList object for our protein values. Let’s investigate the other components of this object:
R
names(y.protein)
OUTPUT
[1] "E" "genes" "other" "targets" "dpc" R
names(y.protein$other)
OUTPUT
[1] "n.observations" "standard.error"R
names(y.protein$genes)
OUTPUT
[1] "Protein.Group" "Protein.Names" "Genes" "NPeptides"
[5] "PropObs" | EList component | Description |
|---|---|
| E | A complete matrix of log2 protein values with no missing data, with samples as columns and proteins as rows |
| genes | Feature annotation dataframe, with columns for: UniProt IDs (Protein.Group), proteins (Protein.Names), gene names (Genes), number of peptides used to quantify each protein (NPeptides), and the proportion of possible observations used to generate each protein value (PropObs) |
| other | List of two matrices with columns and rows corresponding to protein matrix E: number of peptides observed for each protein in each sample (n.observations), and the standard error associated with each protein value for each sample (standard.error) |
| targets | Sample annotation dataframe |
| dpc | DPC intercept and slope coefficient used for protein quantification |
Challenge
Compare the first few rows of the protein (E),
n.observations and standard.error matrices.
Take a look at the NPeptides and PropObs
values for those proteins also. What do you notice about the
relationship between these quality metrics?
Proteins with smaller NPeptides and PropObs
values, and smaller n.observations for individual samples,
have higher standard error.
The more peptide data available for a particular protein in a particular sample, the greater the confidence in the protein value, and therefore the lower the standard error.
If you are interested in a particular protein, it is important to look at these quality metrics for each of your samples.
Dimensionality Reduction and QC
Let’s take a look at our dataset. The plotMDSUsingSEs()
function, or Multidimensional Scaling Plot of Gene Expression
Profiles, Using Standard Errors, is similar to
plotMDS() in the limma package, but takes
account of the standard errors generated by dpcQuant().
This is essentially a PCA plot for the newly generated protein values
that takes into consideration their standard error.
R
plotMDSUsingSEs(y.protein)

That plot is extremely messy and doesn’t tell us much! Let’s clean it up.
R
# Factorise and set colors for class and batch
Class <- factor(y.peptide$targets$Class)
Class.color <- Class
levels(Class.color) <- hcl.colors(nlevels(Class), palette = "cividis")
Batch <- factor(y.peptide$targets$Batch)
Batch.color <- Batch
levels(Batch.color) <- hcl.colors(nlevels(Batch), palette = "Set 2")
R
# Class visualisation
plotMDSUsingSEs(y.protein, pch=16, col=as.character(Class.color))

R
# Batch visualisation
plotMDSUsingSEs(y.protein, pch=16, col=as.character(Batch.color))

R
# Class (labels) and batch (colors) visualisation
plotMDSUsingSEs(y.protein, labels=Class, col=as.character(Batch.color))

We can already see a distinction between the aCD and Ctrl groups, which will be explored further in the next lesson.
It also looks like there are some differences between the batches. We should add batch as a covariate to our analyses in the next lesson.
Managing batch effects
Explaining the different methods of identifying, accounting and correcting for batch effects in proteomics data is an entire tutorial itself! For further information, we recommend the Melbourne Integrative Genomics Workshop: Managing batch effects in biological studies.
If you are concerned about batch effects in your data, University of Melbourne staff and students or affiliates can also contact us for a free consultation.
Normalisation
Normalisation is another key element of the proteomics data cleaning workflow. Normalisation techniques aim to remove non-biological variation in the data, for example, variation due to sample loading or batch effects.
Log transformation (which was automatically applied when we imported our peptide data) helps to normalise intensity values. The default RT-dependent normalisation applied during DIA-NN processing is also quite effective and often sufficient for DIA data; however, each experimental environment and techniques will have a different optimal approach. You may wish to apply an additional method of normalisation to your data.
To learn more about normalisation, keep an eye on the Melbourne Bioinformatics Eventbrite page for a normalisation workshop coming in 2026.
To learn more about different normalisation techniques for proteomics data, we recommend the following papers:
Peng, H., Wang, H., Kong, W., Li, J., & Goh, W. W. B. (2024). Optimizing differential expression analysis for proteomics data via high-performing rules and ensemble inference. Nature communications, 15(1), 3922. https://doi.org/10.1038/s41467-024-47899-w
Tseng, C. Y., Salguero, J. A., Breidenbach, J. D., Solomon, E., Sanders, C. K., Harvey, T., Thornhill, M. G., Palmisano, S. J., Sasiene, Z. J., Blackwell, B. R., McBride, E. M., Luchini, K. A., LeBrun, E. S., Alvarez, M., Mach, P. M., Rivera, E. S., & Glaros, T. G. (2025). Evaluation of normalization strategies for mass spectrometry-based multi-omics datasets. Metabolomics : Official journal of the Metabolomic Society, 21(4), 98. https://doi.org/10.1007/s11306-025-02297-1
Välikangas, T., Suomi, T., & Elo, L. L. (2018). A systematic evaluation of normalization methods in quantitative label-free proteomics. Briefings in bioinformatics, 19(1), 1–11. https://doi.org/10.1093/bib/bbw095
Good work! We now have a clean dataset ready for analysis.
- Key elements of the proteomics data cleaning workflow include: quality filtering, removing contaminants, protein quantification, managing batch effects, and normalisation.
- We can use the
limpapackage in R to clean our peptide data and generate a complete, analysis-ready protein matrix with corresponding standard error values.
Content from Differential expression analysis
Last updated on 2025-11-14 | Edit this page
Overview
Questions
- How can we identify the differentially expressed proteins between our experimental groups?
Objectives
- Conduct differential expression analysis using
limpa - Visualise the top differentially expressed proteins
Differential Expression Analysis (DEA)
Now it is time to analyse our data and investigate proteins that are
differentially expressed between our two experimental groups (control vs
active Crohn’s disease). We will make use of the limma
package and closely related limpa functions specifically
designed for proteomics data.
First, we will fit a linear model that includes both Class and Batch effects.
R
# Set the control group as our reference
Class <- relevel(Class, ref = "Ctrl")
## We also need to reset our colors to match the correct labels
Class.color <- Class
levels(Class.color) <- hcl.colors(nlevels(Class), palette = "cividis")
# Fit a linear model
design <- model.matrix(~Class + Batch)
design
OUTPUT
(Intercept) ClassaCD BatchB3
1 1 1 0
2 1 1 0
3 1 0 0
4 1 1 0
5 1 0 0
6 1 0 0
7 1 1 0
8 1 1 0
9 1 0 0
10 1 0 0
11 1 0 0
12 1 1 1
13 1 0 1
14 1 1 1
15 1 1 1
16 1 0 1
17 1 0 1
18 1 1 1
19 1 0 1
20 1 0 1
attr(,"assign")
[1] 0 1 2
attr(,"contrasts")
attr(,"contrasts")$Class
[1] "contr.treatment"
attr(,"contrasts")$Batch
[1] "contr.treatment"We can see both Class and Batch accounted for in our design matrix.
Now we run the limpa differential expression
analysis function dpcDE() to identify the top
differentially expressed proteins in our dataset.
R
fit <- dpcDE(y.protein, design, plot = TRUE)

R
fit <- eBayes(fit)
# Save our DEA results as a dataframe
results <- topTable(fit, coef = 2, number = Inf)
head(results, n=10)
OUTPUT
Protein.Group Protein.Names Genes NPeptides PropObs logFC
PRTN3_HUMAN P24158 PRTN3_HUMAN PRTN3 11 0.5318182 4.466373
S10A9_HUMAN P06702 S10A9_HUMAN S100A9 23 0.6086957 3.809771
S10A8_HUMAN P05109 S10A8_HUMAN S100A8 17 0.6500000 3.811580
TRFL_HUMAN P02788 TRFL_HUMAN LTF 46 0.3152174 3.998157
CEAM8_HUMAN P31997 CEAM8_HUMAN CEACAM8 6 0.3583333 3.130768
SAMP_HUMAN P02743 SAMP_HUMAN APCS 8 0.5500000 2.720497
A1AG1_HUMAN P02763 A1AG1_HUMAN ORM1 11 0.4454545 3.349033
CAP7_HUMAN P20160 CAP7_HUMAN AZU1 6 0.4666667 3.200933
FRIH_HUMAN P02794 FRIH_HUMAN FTH1 12 0.5625000 2.528228
RNAS2_HUMAN P10153 RNAS2_HUMAN RNASE2 6 0.5666667 2.887339
AveExpr t P.Value adj.P.Val B
PRTN3_HUMAN 11.146376 6.909177 8.925046e-08 2.499013e-05 7.9144156
S10A9_HUMAN 12.809535 6.354519 4.259264e-07 5.962969e-05 6.4365522
S10A8_HUMAN 12.865666 6.070836 9.561924e-07 8.924462e-05 5.6692556
TRFL_HUMAN 9.061431 5.755654 2.361294e-06 1.601370e-04 4.8072839
CEAM8_HUMAN 10.078482 5.689096 2.859590e-06 1.601370e-04 4.6265751
SAMP_HUMAN 11.028982 5.317255 8.351615e-06 3.897420e-04 3.6088844
A1AG1_HUMAN 10.284907 5.228167 1.079921e-05 4.319682e-04 3.3650142
CAP7_HUMAN 10.422828 4.613155 6.335616e-05 2.217466e-03 1.6820861
FRIH_HUMAN 11.341144 4.227543 1.897414e-04 5.396242e-03 0.6493737
RNAS2_HUMAN 11.228811 4.222021 1.927229e-04 5.396242e-03 0.6366061A closer look at the results
Our output table is ordered by B statistic. Run
View(results) to take a closer look at some of the lower
ranked proteins.
Are there any proteins with an absolute fold change > 2 (|logFC| > 1) that are not statistically significant? Can you think of some explanations for this?
Hint: Take note of the PropObs value and remember its relationship to standard error.
Visualise differentially expressed proteins
Expression values and standard error by sample
We can visualise the log2 expression values for individual proteins, together with standard errors.
R
plotProtein(y.protein, "PRTN3_HUMAN", col = as.character(Class.color))
legend('topleft', legend = levels(Class), fill = levels(Class.color))

R
plotProtein(y.protein, "S10A9_HUMAN", col = as.character(Class.color))
legend('topleft', legend = levels(Class), fill = levels(Class.color))

Challenge
This simple function plots the protein values from our expression
matrix, which we did not directly edit to mitigate batch effects. Try
running the limma function to remove batch effects from our
dataset and see how this changes your plot.
R
y.protein.rbe <- y.protein
y.protein.rbe$E <- removeBatchEffect(y.protein,batch = y.protein$targets$Batch, group=y.protein$targets$Class)
plotProtein(y.protein.rbe, "S10A9_HUMAN", col = as.character(Class.color))
legend('topleft', legend = levels(Class), fill = levels(Class.color))

Heatmap
We can filter our results to the up to 50 top most significant differentially expressed proteins, then visualise via a clustered heatmap.
R
# Filter for the up to 50 most significant results
sig_proteins <- results %>%
filter(adj.P.Val < 0.05) %>% # filter by p value
top_n(50, wt = abs(logFC)) %>% # filter by absolute log fold change
pull(Protein.Names)
expr_matrix <- y.protein$E[sig_proteins, ]
# Scale the data and visualise via heatmap
scaled_expr <- t(scale(t(expr_matrix)))
pheatmap(scaled_expr,
cluster_rows = TRUE,
cluster_cols = TRUE,
show_rownames = TRUE,
show_colnames = FALSE,
annotation_col = y.protein$targets,
clustering_method = 'ward.D')

Challenge
How would you interpret this heatmap?
Volcano plot
We can also visualise our results as a volcano plot using
EnhancedVolcano().
R
EnhancedVolcano(results,
lab = results$Genes,
x = 'logFC',
y = 'adj.P.Val',
pCutoff = 0.05,
FCcutoff = 1.0,
pointSize = 2.0,
labSize = 3.5,
drawConnectors = TRUE,
maxoverlapsConnectors = Inf,
lengthConnectors = unit(0.001, 'npc'),
typeConnectors ='closed',
ylim = c(0, 5),
xlim = c(-5, 5))

- We can fit a linear model to our data and conduct differential
expression analysis using
limpaandlimmafunctions. - There are many different ways to visualise differentially expressed proteins.
Content from Network and Enrichment Analysis
Last updated on 2025-11-14 | Edit this page
Overview
Questions
- How can DEA results be used to identify biologically meaningful patterns?
- How can we visualise and interpret protein–protein interaction (PPI) networks and pathway enrichment results?
Objectives
- Visualise significant proteins using network analysis (STRING)
- Identify enriched biological processes and pathways (GO and KEGG)
Now that we have a list of differentially expressed (DE) proteins, we can explore how these proteins interact with each other and what biological functions or pathways they are involved in.
This step helps translate statistical significance into biological meaning by integrating our results with known protein–protein interactions (PPIs) and functional annotation databases.
STRING protein–protein interaction network analysis
The STRING database integrates known and predicted PPIs from multiple sources (e.g. experimental data, text mining, prediction methods, and curated databases).
We can visualise DE proteins within the context of their interaction network to identify clusters or modules potentially corresponding to biological pathways.
R
# Create an instance of the STRINGdb reference class for humans (species 9606)
string_db <- STRINGdb$new(version = "11.5", species = 9606, score_threshold = 400)
# Map STRING identifiers to gene identifiers in our DEA results dataframe
mapped <- string_db$map(results, "Protein.Names", removeUnmappedRows = TRUE)
# Only map the proteins of significance
de_proteins_mapped <- mapped %>% filter(adj.P.Val < 0.05)
# Plot network of significant proteins
string_db$plot_network(de_proteins_mapped$STRING_id)
Note: if the above code takes too long to load, you can download
an RData that contains the string_db and
mapped functions here.

How many clusters are there?
How many of these interactions are predicted versus experimentally validated? (See image below for legend)
You can explore how these proteins interact in an interactive setting by going to https://string-db.org/ and further exploring your proteins of interest.

More data exploration using STRINGdb
There are many ways of exploring your data using the STRING database - too many to cover in one tutorial!
You can learn more by reading the vignette
or inspecting available functions within the STRINGdb
package by running:
R
STRINGdb$methods()
OUTPUT
[1] ".objectPackage" ".objectParent"
[3] "add_diff_exp_color" "add_proteins_description"
[5] "benchmark_ppi" "benchmark_ppi_pathway_view"
[7] "callSuper" "copy"
[9] "enrichment_heatmap" "export"
[11] "field" "get_aliases"
[13] "get_annotations" "get_bioc_graph"
[15] "get_clusters" "get_enrichment"
[17] "get_graph" "get_homologs"
[19] "get_homologs_besthits" "get_homology_graph"
[21] "get_interactions" "get_link"
[23] "get_neighbors" "get_paralogs"
[25] "get_pathways_benchmarking_blackList" "get_png"
[27] "get_ppi_enrichment" "get_ppi_enrichment_full"
[29] "get_proteins" "get_pubmed"
[31] "get_pubmed_interaction" "get_subnetwork"
[33] "get_summary" "get_term_proteins"
[35] "getClass" "getRefClass"
[37] "import" "initFields"
[39] "initialize" "load"
[41] "load_all" "map"
[43] "mp" "plot_network"
[45] "plot_ppi_enrichment" "post_payload"
[47] "ppi_enrichment" "remove_homologous_interactions"
[49] "set_background" "show"
[51] "show#envRefClass" "trace"
[53] "untrace" "usingMethods" Szklarczyk, D., Nastou, K., Koutrouli, M., Kirsch, R., Mehryary, F., Hachilif, R., Hu, D., Peluso, M. E., Huang, Q., Fang, T., Doncheva, N. T., Pyysalo, S., Bork, P., Jensen, L. J., & von Mering, C. (2025). The STRING database in 2025: protein networks with directionality of regulation. Nucleic acids research, 53(D1), D730–D737. https://doi.org/10.1093/nar/gkae1113
Functional enrichment analysis (GO and KEGG)
Using the significant DE proteins, we can perform functional enrichment analysis to determine whether certain biological processes (BP), cellular components (CC), or molecular functions (MF) are over-represented.
Two common types of enrichment exploration are:
- Gene Ontology (GO): describes BPs, CCs, amd MFs
- KEGG pathways: curated molecular interactions
GO enrichment analysis
R
#Subset genes from results table
de_proteins <- results %>% filter(adj.P.Val < 0.05)
# Convert UniProt IDs to Entrez IDs for enrichment analysis
converted <- bitr(de_proteins$Protein.Group,
fromType = "UNIPROT",
toType = "ENTREZID",
OrgDb = org.Hs.eg.db)
OUTPUT
'select()' returned 1:1 mapping between keys and columnsWARNING
Warning in bitr(de_proteins$Protein.Group, fromType = "UNIPROT", toType =
"ENTREZID", : 7.69% of input gene IDs are fail to map...R
# GO Biological Process enrichment
ego <- enrichGO(gene = converted$ENTREZID,
OrgDb = org.Hs.eg.db,
ont = "ALL", # looking at all 3 subontologies
pAdjustMethod = "BH",
pvalueCutoff = 0.05,
readable = TRUE)
# Show plot
dotplot(ego, showCategory = 10)

KEGG pathway enrichment analysis
R
# KEGG pathway enrichment
ekegg <- enrichKEGG(gene = converted$ENTREZID,
organism = 'hsa',
pvalueCutoff = 0.05)
OUTPUT
Reading KEGG annotation online: "https://rest.kegg.jp/link/hsa/pathway"...OUTPUT
Reading KEGG annotation online: "https://rest.kegg.jp/list/pathway/hsa"...R
dotplot(ekegg, showCategory = 10)

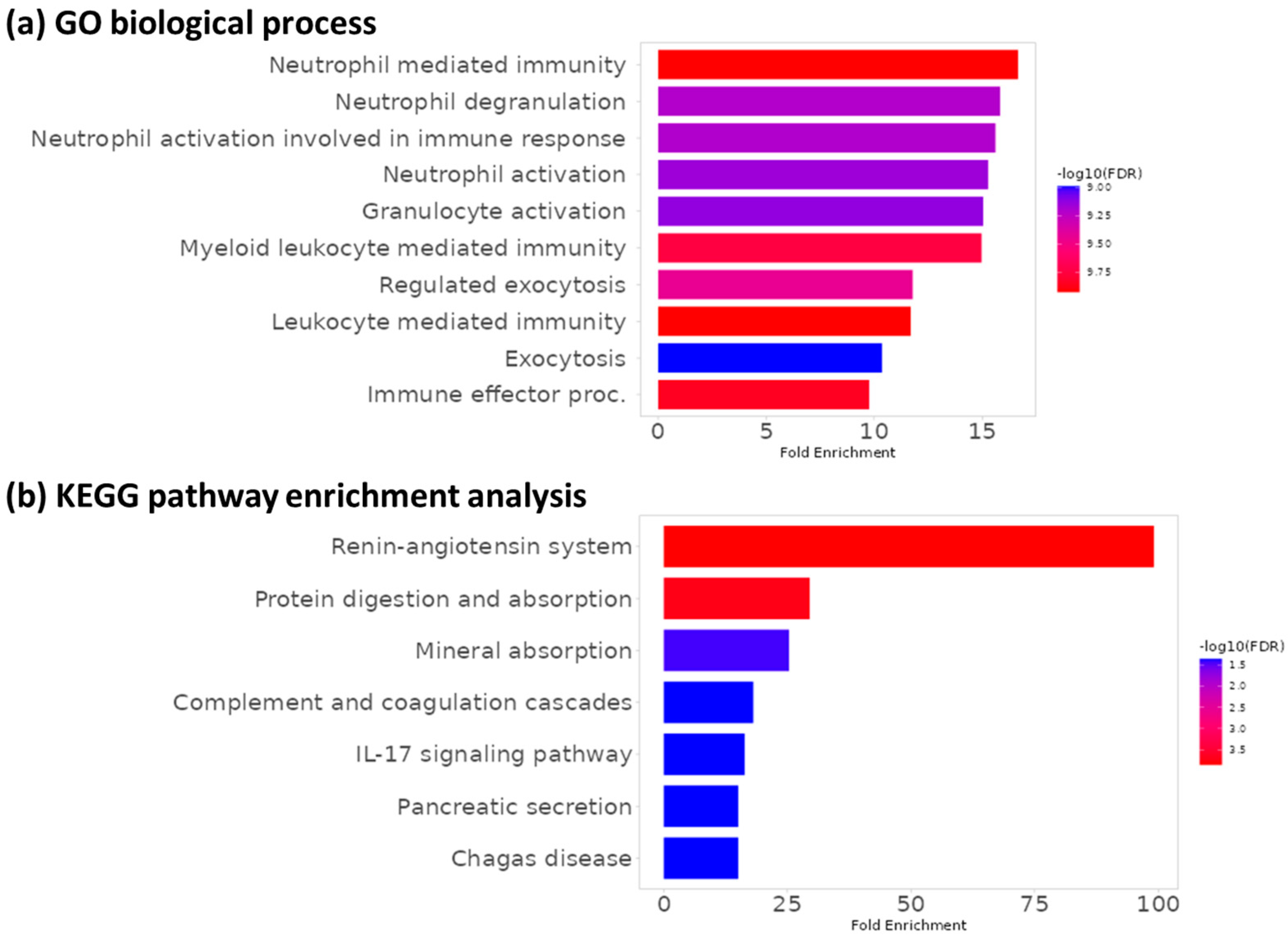{kind=link}
While there are many similar pathways, the exact pathways are not identical. This is likely due to the fact we are using a subset of the larger dataset in this tutorial, so we have a slightly different list of DE proteins.
- Differential expression analysis identifies statistically significant changes in protein abundance between conditions, but does not necessarily tell us the biological significance of such identified proteins.
- Network analysis (STRING) can reveal physical or functional interactions among DE proteins.
- Enrichment analysis (GO/KEGG) can link those proteins to known biological processes or pathways.
- Using multiple approaches can help you interpret proteomics data to measure reliability and consistency of results.